En geometría
y cristalografía las redes de Bravais
son una disposición infinita de puntos discretos cuya estructura es invariante
bajo cierto grupo de traslaciones. En la mayoría de casos
también se da una invariancia bajo rotaciones o simetría rotacional. Estas
propiedades hacen que desde todos los nodos de una red de Bravais se tenga la
misma perspectiva de la red. Se dice entonces que los puntos de una red de
Bravais son equivalentes.
Mediante teoría de grupos se ha demostrado que sólo
existe una única red de Bravais unidimensional, 5 redes bidimensionales y 14
modelos distintos de redes tridimensionales.
La red
unidimensional es elemental siendo ésta una simple secuencia de nodos
equidistantes entre sí. En dos o tres dimensiones
las cosas se complican más y la variabilidad de formas obliga a definir ciertas
estructuras patrón para trabajar cómodamente con las redes.
Para generar
éstas normalmente se usa el concepto de celda
primitiva. Las celdas unitarias, son paralelogramos (2D) o paralelepípedos
(3D) que constituyen la menor subdivisión de una red cristalina que conserva
las características generales de toda la retícula, de modo que por simple
traslación de la misma, puede reconstruirse la red al completo en cualquier
punto.
Una red
típica R en R tiene la forma:

donde {a1,...,
an} es una base en el espacio Rn. Puede haber diferentes bases que generen la
misma red pero el valor absoluto del determinante de los vectores ai vendrá
siempre determinado por la red por lo que se lo puede representar como d(R).
Las celdas unitarias se
pueden definir de forma muy simple a partir de dos vectores (2D) o tres
vectores (3D). La construcción de la celda se realiza trazando las paralelas de
estos vectores desde sus extremos hasta el punto en el que se cruzan. Existe un
tipo de celda unitaria que se construye de un modo distinto y que presenta
ciertas ventajas en la visualización de la red ya que posee la misma simetría
que la red, es la celda de Wigner-Seitz. Una celda unitaria se caracteriza
principalmente por contener un único nodo de la red de ahí el adjetivo de
"unitaria". Si bien en muchos casos existen distintas formas para las
celdas unitarias de una determinada red el volumen de toda celda unitaria es
siempre el mismo.

En
ocasiones resulta más sencillo construir otro tipo de celdas que sin ser
unitarias describen mejor la estructura de la red que tratamos. Este tipo de
celdas se denominan celdas convencionales. Éstas tienen, a su vez, sus propios
parámetros de red y un volumen determinado.
Todas
estas celdas se consideran celdas primitivas ya que son capaces de cubrir todo
el espacio mediante traslaciones sin que queden huecos ni solapamientos. Sus
diferencias o características son las siguientes:
Empaquetamiento compacto:
Esto es cuando los átomos de la celda están en contacto unos con otros. No
siempre será así y en muchos casos mediará una distancia mínima entre las nubes
electrónicas de los diferentes átomos.
Parámetro de red: Es
la longitud de los lados de la celda unitaria. Puede haber tan solo uno, dos o
hasta tres parámetros de red distintos dependiendo del tipo de red de bravais
que tratemos. En las estructuras más comunes se representa con la letra a y con
la c en caso de haber dos.
Nodos o átomos por celda:
Tal y como dice el nombre es el número de nodos o átomos que posee cada celda.
Una celda cuadrada, por ejemplo, poseerá un nodo por celda ya que cada esquina
la comparte con cuatro celdas más. De hecho si una celda posee más de un nodo
de red es que no es unitaria, en cambio si posee más de un átomo por celda
pudiera ser que estuviésemos en una celda unitaria pero con una base atómica de
más de un átomo.
Número de coordinación: Es
el número de puntos de la red más cercanos, los primeros vecinos, de un nodo de
la red. Si se trata de una estructura con empaquetamiento compacto el número de
coordinación será el número de átomos en contacto con otro. El máximo es 12.
Factor de empaquetamiento:
Fracción del espacio de la celda unitaria ocupada por los átomos, suponiendo
que éstos son esferas sólidas.

Donde
f es el factor de empaquetamiento o fracción de volumen ocupado, n el número de
átomos por celda, v el volumen del átomo y Vc el volumen de la celda.
Normalmente se suele dar el factor de empaquetamiento compacto para las
diferentes celdas como indicador de la densidad de átomos que posee cada
estructura cristalina. En este caso los átomos se tratan como esferas rígidas
en contacto con sus vecinos más cercanos.
Densidad: A
partir de las características de la red, puede obtenerse la densidad teórica
del material que conforma la red mediante la siguiente expresión.
Donde
ρ es la densidad, NA el número de Avogadro y m la masa atómica.

EJEMPLO DENSIDAD
VOLUMETRICA:
El
hierro tiene una estructura cristalina BCC y un radio atómico de 1,24 A° a
temperatura ambiente. Calcule el valor teórico de la densidad del hierro y
compare su resultado con el valor experimental 7,87 g/cm3. El peso atómico del
hierro es 55,85 UAM. G/mol
(BCC)
= estructura cristalina cubica centrada en el cuerpo)
Solución:
(4r)2 = 2a2 +
a2 Para
la celda unidad BCC ,
donde
a es la dimensión del lado del cristal, y r es el radio atómico de Fe

Densidad
volumétrica del cobre = rv = masa/volumen
En
la celda unidad hay dos átomos (1/8*8+1 = 2 átomos/celda unitaria). Cada átomo
de hierro tiene una masa de (55,85 UAM/6,02 x 1023 UAM/g)= 9,277*1023 (g/átomo).
volumen de la celda de
unidad del Fe es:

La
densidad del hierro es:

El
resultado es un poco mayor que el experimental ya que no considera los defectos
(huecos) del material real.
Volumen de la celda unitaria
primitiva: Toda celda unitaria tiene el mismo volumen representado
por la siguiente fórmula.

Donde
a son los vectores de la base de la red.
Índice de Miller:
Para
poder identificar unívocamente un sistema de planos cristalográficos se les
asigna un juego de tres números que reciben el nombre de índices de Miller. Los
índices de un sistema de planos se indican genéricamente con las letras (h k
l).
Los
índices de Miller son números enteros, negativos o positivos, y son primos
entre sí. El signo negativo de un índice de Miller debe ser colocado sobre
dicho número.
El
índice de Miller fue presentado por primera vez por el mineralogista británico
William Hallowes Miller en 1839. Existen además otras notaciones1 para los
casos especiales de cristales con planos simétricos

Redes tridimensionales
En función de los parámetros de la celda
unitaria, longitudes de sus lados y ángulos que forman, se
distinguen 7 sistemas cristalinos.
Para
determinar completamente la estructura cristalina elemental de un sólido,
además de definir la forma geométrica de la red, es necesario establecer las
posiciones en la celda de los átomos o moléculas que forman el sólido cristalino; lo que se denominan
puntos reticulares. Las alternativas son las siguientes:
- P: Celda primitiva o simple en
la que los puntos reticulares son sólo los vértices del paralelepípedo.
- F: Celda centrada en las caras,
que tiene puntos reticulares en las caras, además de en los vértices. Si
sólo tienen puntos reticulares en las bases, se designan con las letras A,
B o C según sean las caras que tienen los dos puntos reticulares.
- I: Celda centrada en el cuerpo
que tiene un punto reticular en el centro de la celda, además de los
vértices.
- C: Primitiva con ejes iguales y
ángulos iguales ó hexagonal doblemente centrada en el cuerpo, además de
los vértices.
Combinando
los 7 sistemas cristalinos con las disposiciones de los puntos de red
mencionados, se obtendrían 28 redes cristalinas posibles. En realidad, como
puede demostrarse, sólo existen 14 configuraciones básicas, pudiéndose el resto
obtener a partir de ellas. Estas estructuras se denominan redes de Bravais.

Base atómica
En
el caso más sencillo, a cada punto de red le corresponderá un átomo, pero en
estructuras más complicadas, como materiales cerámicos y compuestos, cientos de
átomos pueden estar asociados a cada punto de red formando celdas unitarias
extremadamente complejas. La distribución de estos átomos o moléculas
adicionales se denomina base atómica y esta nos da su distribución dentro de la
celda unitaria.
Existen
dos casos típicos de bases atómicas. La estructura del diamante y la hexagonal
compacta. Para redes bidimensionales un caso ejemplar sería el grafito cuya
estructura sigue un patrón de red en panal.

Los
nombres (BCC, HCP, FCC) están en nomenclatura internacional o inglesa, los nombres
en la nomenclatura española serían:
"CS"
Cúbica Simple
"BCC"
Cúbica Centrada en el Cuerpo
"FCC"
Cúbica Centrada en las Caras
"HC"
Hexagonal Compacta
Caracterizar la estructura
cristalina de los materiales a partir de los parámetros reticulares.
Ley de Bragg
La ley de Bragg permite estudiar las direcciones en las que la difracción
de rayos X
sobre la superficie de un cristal produce interferencias constructivas, dado que
permite predecir los ángulos en los que los rayos X son difractados por un
material con estructura atómica periódica (materiales cristalinos).
Fue derivada por los físicos
británicos William Henry Bragg y su hijo William Lawrence Bragg en 1913. La ley de Bragg
confirma la existencia de partículas reales en la escala atómica,
proporcionando una técnica muy poderosa de exploración de la materia, la difracción de rayos X. Los Bragg fueron
galardonados con el Premio Nobel de Física en 1915 por sus trabajos
en la determinación de la estructura cristalina del NaCl,
el ZnS y el diamante.
Interferencia
y difracción
Cuando los rayos X alcanzan
un átomo interactúan con sus electrones exteriores. Estos reemiten la radiación
electromagnética incidente en diferentes direcciones y con la misma frecuencia
(en realidad debido a varios efectos hay pequeños cambios en su frecuencia).
Este fenómeno se conoce como dispersión de Rayleigh (o dispersión
elástica). Los rayos X reemitidos desde átomos cercanos interfieren entre sí
constructiva o destructivamente. Este es el fenómeno de la difracción.
En el diagrama que sigue se
esquematizan rayos X que inciden sobre un cristal. Los átomos superiores
reemiten la radiación tras ser alcanzados por ella. Los puntos en los que la
radiación se superpone constructivamente se muestran como la zona de
intersección de los anillos. Se puede apreciar que existen ángulos
privilegiados en los cuales la interferencia es constructiva, en este caso
hacia la derecha con un ángulo en torno a 45º.

La
radiación incidente llega a átomos consecutivos con un ligero desfase
(izquierda). La radiación dispersada por los átomos (círculos azules)
interfiere con radiación dispersada por átomos adyacentes. Las direcciones en
las que los círculos se superponen son direcciones de interferencia
constructiva.
Ley
de Bragg
La interferencia es constructiva
cuando la diferencia de fase entre la radiación emitida por diferentes átomos
es proporcional a 2π. Esta condición se expresa en la ley de Bragg:

Donde
- n es un número
entero,
- λ es la longitud de onda de los rayos X,
- d es la distancia
entre los planos de la red cristalina y,
- θ es el ángulo
entre los rayos incidentes y los planos de dispersión.

De
acuerdo al ángulo de desviación (2θ), el cambio de fase de las ondas produce
interferencia constructiva (figura izquierda) o destructiva (figura derecha).
Deducción
de la Ley de Bragg
Consideramos la figura de
abajo conformada por planos de átomos distanciados a una longitud d. Para el
primer plano, las rayos 1 y 1a golpean los átomos K y P los cuales son
dispersados en todas la direcciones; pero para cierta dirección, estos rayos
(1’ y 1a') se encuentran en fase y por lo tanto se cumple que:
Esta condición se cumple para
cada plano.
Deducción de Ley de Bragg por diferencia de camino
óptico.
Para analizar los rayos
dispersados por átomos en diferentes planos se toma los rayos 1 y 2 de la
figura de arriba. Estos rayos son dispersados por los átomos K y L, la
diferencia en sus caminos ópticos es:

Así estos rayos estarán
completamente en fase si su diferencia de caminos es igual a un número entero
(n) de longitudes de onda  , de tal manera que se cumple que:
, de tal manera que se cumple que:
Otra manera de deducir la Ley
de Bragg es considerar ahora una diferencia de fase. Para dos rayos difractados
se tiene que la diferencia de fase es igual:

Donde , r es la
distancia de separación entre los planos y K es el vector de onda. Para la Fig.
de arriba,

Para que haya una
interferencia constructiva r R es un múltiplo de uno de tal manera que:
Analogía
Se puede expresar esta ley
considerando una analogía con un caso más simple. Consideremos que los planos
cristalográficos son representados por espejos semi transparentes en los que la
radiación incidente es reemitida en parte en cada uno de los planos. Las
interferencias formadas entonces se rigen por la ley de Bragg. De hecho, la
fórmula de Bragg es idéntica a las interferencias producidas en una capa
delgada de aire obtenidas en un interferómetro
de Michelson. De manera más estricta hay que tener en cuenta que las ondas son
dispersadas por átomos individuales alineados de manera periódica.

Interferencias
producidas en una capa delgada de aire. Analogía con la ley de Bragg.






IMPERFECCIONES EN LAS REDES
CRISTALINAS:
Las
imperfecciones se encuentran dentro de la zona de ordenamiento de largo alcance
(grano) y se clasifican de la siguiente manera:
DEFECTOS PUNTUALES (puntos
defectuosos):
Defectos
puntuales: Los defectos puntuales son discontinuidades de la red que involucran
uno o quizá varios átomos. Estos defectos o imperfecciones,, pueden ser
generados en el material mediante el movimiento de los átomos al ganar energía
por calentamiento; durante el procesamiento del material; mediante la introducción
de impurezas; o intencionalmente a través de las aleaciones.
Huecos: Un
Hueco se produce cuando falta un átomo en un sitio normal. Las vacancias se
crean en el cristal durante la solidificación a altas temperaturas o como
consecuencia de daños por radiación. A temperatura ambiente aparecen muy pocas
vacancias, pero éstas se incrementan de manera exponencial conforme se aumenta
la temperatura.
Defectos intersticiales: Se
forma un defecto intersticial cuando se inserta un átomo adicional en una
posición normalmente desocupada dentro de la estructura cristalina. Los átomos
intersticiales, aunque mucho más pequeños que los átomos localizados en los
puntos de la red, aún así son mayores que los sitios intersticiales que ocupan;
en consecuencia, la red circundante aparece comprimida y distorsionada. Los
átomos intersticiales como el hidrógeno a menudo están presentes en forma de
impurezas; los átomos de carbono se agregan al hierro para producir acero. Una
vez dentro del material, el número de átomos intersticiales en la estructura se
mantiene casi constante, incluso al cambiar la temperatura.
Defectos sustitucionales: Se
crea un defecto sustitucional cuando se remplaza un átomo por otro de un tipo
distinto. El átomo sustitucional permanece en la posición original. Cuando
estos átomos son mayores que los normales de la red, los átomos circundantes se
comprimen; si son más pequeños, los átomos circundantes quedan en tensión. En
cualquier caso, el defecto sustitucional distorsiona la red circundante.
Igualmente, se puede encontrar el defecto sustitucional como una impureza o
como un elemento aleante agregado deliberadamente y, una vez introducido, el
número de defectos es relativamente independiente de la temperatura.
IMPORTANCIA DE LOS DEFECTOS
PUNTUALES: Los defectos puntuales alteran el arreglo
perfecto de los átomos circundantes, distorsionando la red a lo largo de quizás
cientos de espaciamientos atómicos, a partir del defecto. Una dislocación que
se mueva a través de las cercanías generales de un defecto puntual encuentra
una red en la cual los átomos no están en sus posiciones de equilibrio. Esta
alteración requiere que se aplique un esfuerzo más alto para obligar a que la
dislocación venza al defecto, incrementándose así la resistencia del material.
Defectos Lineales ( Dislocaciones):
Las dislocaciones son imperfecciones lineales en una
red que de otra forma sería perfecta. Generalmente se introducen en la red
durante el proceso de solidificación del material o al deformarlo. Aunque en
todos los materiales hay dislocaciones presentes, incluyendo los materiales
cerámicos y los polímeros, son de particular utilidad para explicar la
deformación y el endurecimiento de los metales. Podemos identificar dos tipos
de dislocaciones: la dislocación de tornillo y la dislocación de borde.
Dislocación de tornillo:
La dislocación de tornillo se puede ilustrar haciendo
un corte parcial a través de un cristal perfecto, torciéndolo y desplazando un
lado del corte sobre el otro la distancia de un átomo.
Una dislocación de borde se puede ilustrar haciendo un
corte parcial a través de un cristal perfecto, separándolo y rellenando
parcialmente el corte con un plano de átomos adicional. El borde inferior de
este plano adicional representa la dislocación de borde.
Las dislocaciones mixtas tienen componentes tanto de
borde como de tornillo, con una región de transición entre ambas. El vector de
Burgers, sin embargo, se conserva igual para todas las porciones de la
dislocación mixta.
Aunque en algunos materiales cerámicos y polímeros
puede ocurrir deslizamiento, el proceso de deslizamiento es de particular
utilidad para entender el comportamiento mecánico de los metales.
En primer término, el deslizamiento explica por qué la
resistencia de los metales es mucho menor que el valor predecible a partir del
enlace metálico. Si ocurre el deslizamiento, sólo es necesario que se rompa en
algún momento una pequeña fracción de todas las uniones metálicas a través de
la interfase, por lo que la fuerza requerida para deformar el metal resulta
pequeña.
Segundo, el deslizamiento le da ductilidad a los
metales. Si no hay dislocaciones presentes, una barra de hierro sería frágil;
los metales no podrían ser conformados utilizando los diversos procesos, que
involucran la deformación para obtener formas útiles, como la forja.
En tercer lugar, controlamos las propiedades mecánicas
de un metal o aleación al interferir el movimiento de las dislocaciones. Un
obstáculo introducido en el cristal impedirá que en una dislocación se deslice,
a menos que apliquemos mayor fuerza. Si es necesario aplicar una fuerza
superior, entonces el metal resulta ser más resistente. Estos obstáculos pueden
ser defectos puntuales o borde de grano.
En cuarto lugar, se puede prevenir el deslizamiento de
las dislocaciones achicando el tamaño de grano o introduciendo átomos de
diferente tamaño, que son las aleaciones.
En los materiales se encuentran enormes cantidades de
dislocaciones. La densidad de dislocaciones, o la longitud total de
dislocaciones por unidad de volumen, generalmente se utiliza para representar
la cantidad de dislocaciones presentes. Densidades de dislocación de 10ˆ-6
cm/cm3 son típicas en los metales más suaves, en tanto que se pueden conseguir
densidades de hasta 10ˆ-12 cm/cm3 deformando el material.
|
La estructura final resultante está constituida por un agrupamiento de granos o cristales de forma irregular pero guardando cada uno una orientación fija y bien determinada. |
 |
Los defectos de superficie son las fronteras o planos
que separan un material en regiones de la misma estructura cristalina pero con
orientaciones cristalográficas distintas, y la superficie externa de un
material. En las superficies externas del material la red termina de manera
abrupta. Cada átomo de la superficie ya no tiene el mismo número de
coordinación y se altera el enlace atómico. Asimismo, la superficie puede ser
muy áspera, contener pequeñas muescas y quizá ser mucho más reactiva que el
interior del material. En líquidos, los átomos en la superficie tienen mayor
energía porque no tienen todos sus átomos vecinos. Esto hace que al tratar de
minimizar la energía se tiende a reducir el numero de átomos en esta condición,
por lo tanto tienden a reducir la superficie respecto al volumen, esto
geométricamente corresponde a una esfera.
|
El crecimiento de los cristales que se inicia en los
centros o núcleos de cristalización en el metal líquido, no puede ser
uniforme a causa de los diferentes factores de la composición del metal, la
velocidad de enfriamiento y las interferencias que se producen entre ellos
mismos durante el proceso de crecimiento.
|
 |
La microestructura de la mayor parte de los materiales
está formada por muchos granos. Un grano es una porción del material dentro del
cual el arreglo atómico es idéntico. Sin embargo, la orientación del arreglo
atómico, o de la estructura cristalina, es distinta para cada grano. En la
figura se muestran de manera esquemática tres granos; la red de cada uno de
ellos es idéntica pero están orientados de manera distinta. La frontera de
grano, que es la superficie que separa los granos, es una zona estrecha en la
cual los átomos no están correctamente espaciados. Esto quiere decir que, en
algunos sitios, los átomos están tan cerca unos de otros en la frontera de
grano que crean una región de compresión y en otras áreas están tan alejados
que crean una región de tensión.
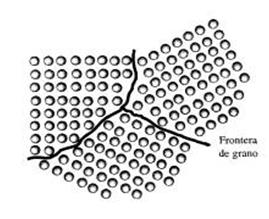
Los átomos cerca de las fronteras de los tres granos
no tienen un espaciamiento o arreglo de equilibrio.
Un método para controlar las propiedades de un
material es controlando el tamaño de los granos. Reduciendo el tamaño de éstos
se incrementa su número y, por tanto, aumenta la cantidad de fronteras de
grano. Cualquier dislocación se moverá solamente una distancia corta antes de
encontrar una frontera de grano, incrementando así la resistencia del metal. Se
puede relacionar el tamaño de grano con el tensión de fluencia del material.
Los límites de grano tienen una influencia importante sobre las propiedades del
metal, su número y tamaño esta en función de la tasa de Nucleacion y los índices
de crecimiento de este. Una vez que el metal se ha solidificado, se puede
modificar el tamaño y numero de granos, ya sea por deformación o tratamiento térmico,
lo cual permitirá que sus propiedades mecánicas varíen considerablemente, la
siguiente ecuación muestre su influencia en el esfuerzo de cadencia:
ß = K1 + K2/ D ˆ- 2
Donde ß es el esfuerzo de Cadencia;
K1 y K2 ctes del Material;
D es el tamaño del Grano
Los metales puros y los Eutécticos, solidifican a
temperatura constante, la solidificación se inicia cuando el metal liquido se enfría
hasta su punto de solidificación, luego la temperatura se mantiene uniforme
hasta que la solidificación concluye, mientras esta transformación ocurre el
calor latente de solidificación que desprende el metal, mantiene la temperatura
constante. Si este se enfriase en completa uniformidad y estuviese exento de
impurezas de cualquier índole, podría generarse una cristalización a partir de
cristales al azar dentro del liquido(Nucleacion)

Se necesitan dos condiciones para el crecimiento del
sólido primero, que el crecimiento requiere que el calor latente de fusión, que
se disipa durante la solidificación del líquido, sea eliminado de la interface
sólido líquido. Segundo, y a diferencia de los metales puros, debe ocurrir la
difusión tal de manera que durante el enfriamiento las composiciones de las
fases sólida y líquida sigan las curvas de sólidas y de líquidas. El calor
latente de fusión es eliminado a lo largo de un rango de temperaturas, y así a
curva de enfriamiento muestra un cambio en pendiente, en vez de una meseta
plana. Para poder conseguir esta estructura final en equilibrio, la velocidad
de enfriamiento debe ser extremadamente lenta. Debe permitirse el tiempo
suficiente para que los átomos de primero y el segundo se difundan y produzcan
las composiciones equilibradas. En la mayor parte de las situaciones prácticas,
la velocidad de enfriamiento es demasiado rápida para permitir este equilibrio.

Estructura Cristalina
La
Física del estado sólido constituye una parte importante de la Física Cuántica. Con su ayuda podemos
comprender las propiedades mecánicas, térmicas, eléctrico-magnéticas y ópticas
propias de los sólidos.
La
existencia de la materia en un estado u otro depende de las condiciones de presión
y temperatura
en las que se formaron. De la misma forma, estos parámetros condicionan la
formación de la estructura interna del sólido.
Cada
elemento tiene sus propias curvas de cambio de fase, de manera que dependiendo
del elemento se necesitarán unas condiciones u otras para la formación del
sólido o para realizar cualquier otro cambio de fase.
Dependiendo del alcance del orden espacial de la estructura interna en la
materia y su distribución en la misma podemos distinguir entre:
- Monocristal: Presenta una
fuerte interacción entre sus componentes los cuales describen una mínima
oscilación con poca energía potencial. Las partículas están dispuestas de
acuerdo a un orden en el espacio que está determinado de acuerdo con una
red estructural formada por la "recreación" geométrica de
la celdilla unidad en
toda la estructura del sólido. Presentan lo que se conoce como Anisotropía.
- Policristal: Está compuesto
por diversas regiones en las que individualmente se recrea un monocristal
aunque las disposiciones de cada una de estas regiones no son simétricas
entre sí. Presenta lo que se llama Isotropía estadística.
- Amorfos: No presentan
una estructura o distribución en el espacio, lo cual los determina como
una estructura espacial tridimensional no definida. No se trata de una
estructura cristalina.

Formas y propiedades del
cristal..
En
un modelo de sólido en el que los átomos están conectados entre sí mediante una
especie de "muelles" (los cuales representarían la energía
potencial que los une), la energía interna del sólido se compone de energía
potencial elástica y energía cinética de sus átomos. La
presión es una medida del grado de compresión de sus átomos y la temperatura
una medida de la energía cinética interna del conjunto de los mismos. Esto nos
permite determinar que de acuerdo con las características externas del medio en
que se encuentre, permitirán al elemento en cuestión poder adoptar un estado u
otro e incluso formar o no una estructura cristalina.
Sin
embargo la formación de una estructura cristalina no es un proceso fijo en un
mismo elemento, ya que incluso tratándose así las
condiciones de formación del sólido podrían determinar dos estructuras
cristalinas diferentes para un mismo elemento, la cual otorga las propiedades
tanto físicas y eléctricas como ópticas al nuevo sólido formado. Por ejemplo,
el carbono puede cristalizar en grafito en determinadas condiciones y en otras cristaliza en
el diamante, sin duda las características de uno frente a otro difieren
bastante para tratarse en ambos casos de carbono cristalizado.
Este
proceso no sólo es dependiente de la presión y la temperatura en sí mismos,
sino también del tiempo aplicado en cada uno de dichos factores. De esta forma
se sabe que la formación de cristales requiere un calentamiento del material a
alta temperatura, aproximadamente 200 °C, lo que se conoce como temperatura de cristalización, a
partir de la cual el elemento se funde para posteriormente, después de un
tiempo lo suficientemente largo, cristalice. Al añadir temperatura al material,
realmente le estamos damos energía, permitiendo que las partículas que lo
componen oscilen a mayor velocidad con una mayor energía térmica, logrando que se funda (cambie al estado líquido).
Luego mediante un enfriamiento lento conseguimos dar tiempo a las partículas
que, de forma natural, tienden a retomar una forma geométrica y ordenada en la
red interna consiguiendo así que se forme un cristal.
De
igual forma, si repetimos el proceso pero aplicando un tiempo de enfriamiento
demasiado corto impedimos que las partículas pueda "re-colocarse" en
una red cristalina homogénea haciendo así que la solidificación de lugar a un
amorfo.
El
policristal es el caso más típico de los que puedan encontrarse en la
naturaleza, ya que un monocristal es un caso que rara vez se da. Un cristal
posee diferentes zonas que no pueden homogeneizarse entre si, pero se puede
hacer que sean como monocristales individuales en cada una de sus regiones.
Siguiendo
el ejemplo del carbono, la cualidad de que un mismo elemento pueda cristalizar
en diferentes formas nos lleva al hecho de que es la red cristalina que forman
la que determina sus propiedades. En la naturaleza existen 14 tipos de redes
cristalinas (otras más complejas son combinaciones de estas más simples) que son
conocidas como Redes de Bravais.
Estas
redes son organizaciones geométricas tridimensionales en el espacio
características de las partículas del sólido. Así pueden estudiarse las
distribuciones en la red de los elementos.
Por
ejemplo: El fósforo(P) cristaliza en una estructura cúbica, el hierro (Fe) en
una bcc y la plata (Ag) en una fcc .Otros cristalizan en redes compuestas como
por ejemplo los elementos del grupo IV(C, Si, Ge...) o del III de la tabla periódica que lo hacen en
una estructura de tipo diamante,
que es la combinación de dos redes fcc con una distancia interatómica de 1/4 de
la diagonal.
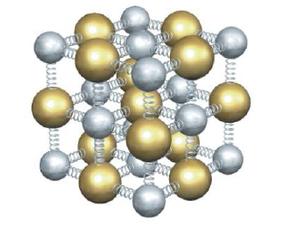
Representación de una
celdilla en la disposición geométrica de sus partículas.
Según
cada una de estas distribuciones, cada una de las partículas situadas en los
nodos de la estructura, contribuye en una parte a la formación del número de
átomos contenido en su interior. Se trata del número de partículas por celdilla elemental que puede
obtenerse como:
Siendo "nv" el
número de partículas en los vértices, "ni" en el interior y
"nf" en las caras del tetraedro.
Debido
a que muchos de los compuestos elementales presentan simetría esférica podemos
visualizarlas considerando éstas como empaquetamientos espaciales de esferas
rígidas. Partiendo de esta idea, podemos determinar la llamada Fracción de
Empaquetamiento que nos proporciona una medida de lo "llena" que
está la estructura reticular:

Para
observar la estructura interna que posee un cristal generalmente puede
determinarse a partir del análisis de la difracción ondulatoria producida
cuando los fotones inciden en el cristal.
Gracias
a estas observaciones W.L.Bragg propuso la conocida Ley de Bragg, que permite ver superficialmente la posición de los
planos que forman los átomos.. Ley de
Bragg:
Estas
mismas propiedades pertenecientes a los sólidos cristalinos y el fundamento de
los cambios de fase es el utilizado en el proceso de grabación de CD-RW y DVD-RW mediante
cambios en la estructura cristalina, haciendo zonas amorfas o policristalinas, según
los datos (bits) que se desean grabar. Otras propiedades y teorías están
relacionadas con la física de los cristales como las bandas de energías o los modelos que explican las
propiedades eléctricas de conductores metálicos y semiconductores.
Modelo de bandas energéticas
El
comportamiento de los electrones está regido por las leyes de la mecánica cuántica, por lo tanto:
- Los
electrones no pueden tener cualquier nivel de energía: los estados de
energía están cuantificados. A un conjunto de niveles de energía muy cerca
entre sí se lo denomina banda de energía y se la considera
continua.
- No
todas las bandas se ocupan uniformemente, sino que algunas tienen más
probabilidades de ser ocupadas que otras, incluso hay bandas totalmente
desocupadas, o sea que la probabilidad de que un electrón tenga ese nivel
de energía es nula o muy cercana a cero.
Véase
también: Modelo de Kronig-Penney.
Modelo
simple
El
modelo
de Drude permitía explicar el comportamiento como conductor de algunos
sólidos basándose en la aplicación de la teoría cinética a los electrones en un
sólido. Sin embargo este modelo era insuficiente a la hora de explicar el
comportamiento de otros materiales que hoy día se conocen como semiconductores.
En respuesta al modelo de Drude surgió el modelo de bandas energéticas, el cual
basándose en las distribuciones de los electrones en sus orbitales a modo de
regiones discretas, puede explicar el comportamiento de la conductividad en los materiales.
Usualmente,
se presenta este esquema basado en el modelo atómico de Bohr y el principio de exclusión de Pauli.
Supóngase
una red cristalina formada por átomos de silicio. Cuando
los átomos están aislados, el orbital s (2 estados
con dos electrones) y el orbital
p (6 estados con 2 electrones y cuatro vacantes) tendrán una cierta energía
Es y Ep respectivamente (punto A). A medida que disminuye
la distancia interatómica comienza a observarse la interacción mutua entre los
átomos, hasta que ambos orbitales llegan a formar, por la distorsión creada, un
sistema electrónico único. En este momento se tienen 8 orbitales híbridos sp³
con cuatro electrones y cuatro vacantes (punto B).
Si
se continúa disminuyendo la distancia interatómica hasta la configuración del
cristal, comienzan a interferir los electrones de las capas internas de los
átomos, formándose bandas de energía (punto Z). Las tres bandas de valores que
se pueden distinguir son:
- Banda de valencia. 4 estados, con
4 electrones.
- Banda
prohibida.
No puede haber electrones con esos valores de energía en el cristal.
-
Banda de conducción. 4 estados, sin
electrones.

Distribución
probabilística de los electrones en las bandas
Los
electrones no se distribuyen uniformemente en las diferentes bandas, sino que
algunas son más probables a ser ocupadas que otras. La probabilidad de
ocupación de las bandas está dada por la estadística de Fermi-Dirac, y el parámetro
más importante es la energía de Fermi.
Conductividad eléctrica
La
conducción eléctrica en un sólido se presenta cuando el mismo tiene
parcialmente llena su banda de conducción. También hay conducción eléctrica
cuando la banda de conducción está vacía y además ésta se traslapa con la banda
de valencia.
Constante de disociación ácida

El ácido acético, un ácido
débil puede perder un protón (destacado en verde) y donarlo a una molécula
de agua, H20,
dando lugar a un anión
acetato CH3COO- y creando un catión hidronio
H3O . Esta es una reacción de equilibrio, por lo que el proceso
inverso también tiene lugar. Rojo: oxígeno, negro: carbono, blanco: hidrógeno.
Una constante de disociación ácida, Ka, (también
conocida como constante de acidez,
o constante de ionización ácida)
es una medida cuantitativa de la fuerza
de un ácido en solución. Es la constante de equilibrio de una reacción
conocida como disociación en el contexto de las reacciones ácido-base. El equilibrio puede
escribirse simbólicamente como:
HA
A- + H+
donde HA es un ácido genérico que se disocia dividiéndose en A-, conocida como base
conjugada del ácido, y el ion
hidrógeno o protón, H+,
que, en el caso de soluciones acuosas, existe como un ion
hidronio solvatado. En el ejemplo que se muestra en la figura, HA representa el ácido acético, y A- el ion acetato. Las
especies químicas HA, A- y H+ se dice que están en
equilibrio cuando sus concentraciones no cambian con el paso del tiempo. La
constante de disociación se escribe normalmente como un cociente de las
concentraciones de equilibrio (en mol/L),
representado por [HA], [A-] y [H+]:

Debido a los muchos órdenes
de magnitud que pueden abarcar los valores de K a,
en la práctica se suele expresar la constante de acidez mediante una medida logarítmica de la constante de acidez, el pKa,
que es igual al -log10, y que también suele ser denominada constante
de disociación ácida:
PKa = -Log 10 Ka
A mayor valor de pKa,
la extensión de la disociación es menor. Un ácido débil
tiene un valor de pKa en un rango aproximado de −2 a 12 en
agua. Los ácidos con valores de pKa menores que
aproximadamente −2 se dice que son ácidos
fuertes; un ácido fuerte está casi completamente disociado en
solución acuosa, en la medida en que la concentración del ácido no disociado es
indetectable. Los valores de pKa para los ácidos fuertes,
pueden ser estimados por medios teóricos o por extrapolación de medidas en
medios no acuosos, en los que la constante de disociación es menor, tales como acetonitrilo
y dimetilsulfóxido.
Definiciones
De
acuerdo con la definición original de Arrhenius, un ácido es una sustancia que se disocia
en solución acuosa, liberando el ion hidrógeno H+ (un
protón):1
HA  A-
+ H+
A-
+ H+
La
constante de equilibrio para esta reacción de disociación se conoce como constante de disociación. El protón
liberado se combina con una molécula de agua para dar un H3O+, y así más
tarde Arrhenius propuso que la disociación se debe escribir como una reacción ácido-base:
HA +
H2O A-
+ H3O+
La
Teoría de Brønsted–Lowry generaliza esto algo
más al considerarla como una reacción de intercambio de protones:
Ácido
+ base base
conjugada + ácido conjugado
El
ácido pierde un protón, dando la base conjugada, el protón se transfiere a la
base, dando el ácido conjugado. Para soluciones acuosas de un ácido HA, la base
es el agua, la base conjugada del ácido es A- y el ácido conjugado
de la base es el ion hidronio. La definición de Brønsted-Lowry se aplica a
otros disolventes, como el dimetilsulfóxido: el solvente S actúa como una
base, aceptando un protón y formando el ácido conjugado SH+.
En
química en solución es común utilizar H+ como una abreviatura del
ion hidrógeno solventado, independientemente del disolvente. En solución
acuosa, H+ denota un ion hidronio sulfatado en lugar de un protón.
La
designación de un ácido o una base como "conjugado" depende del
contexto. El ácido conjugado BH+ de una base B se disocia según la
ecuación:
BH+
+ OH- B +
H2O
que
es la inversa del equilibrio:
H2O
(ácido) + B (base) OH−
(base conjugada) + BH+ (ácido conjugado)
El
ion hidróxido
OH−, una base bien conocida, actúa aquí como la base conjugada del
ácido agua.
Los ácidos y las bases se consideran así simplemente como donadores o aceptores
de protones, respectivamente.
El
agua es anfiprótica: puede reaccionar tanto como un
ácido o como una base. Otro ejemplo de molécula anfiprótica es el ion bicarbonato HCO3- que
es la base conjugada del ácido carbónico H2CO3 en
el equilibrio:
H2CO3
+ H2O HCO3-
+ H3O+
Pero
también el ácido conjugado del ion carbonato CO32- en la reacción
inversa del equilibrio:
HCO3-
+ OH- CO32-
+ H2O
El
equilibrio del ácido carbónico es importante para la hemostasis ácido-base en
el cuerpo humano.
Una
definición más amplia de la disociación ácida incluye la hidrólisis,
en la que los protones son producidos por la separación de moléculas de agua.
Por ejemplo, el ácido bórico (B(OH)3) actúa como un
ácido débil, aunque no es un donante de protones, debido a la hidrólisis:
B(OH)3
+ 2 H2O B(OH)4-
+ H3O+
Del
mismo modo, la hidrólisis de los iones metálicos produce iones como el [Al(H2O)6]3+
que se comportan como ácidos débiles:7
[Al(H2O)6]3+
+ H2O [Al(H2O)5(OH)]2+
+ H3O+
Constante de equilibrio
Una
constante de disociación ácida es un ejemplo particular de una constante de equilibrio. Para el
equilibrio específico entre un ácido monoprótico, HA y su
base conjugada A- en agua:
HA +
H2O A-
+ H3O+
la constante de equilibrio
termodinámica, K puede definirse por:
puede definirse por:

donde
{A} es la actividad de la especie química A, etc. K es adimensional
ya que la actividad no tiene dimensiones . Las actividades de los productos de
disociación están colocados en el numerador, las actividades de los reactivos
están colocadas en el denominador. Ver coeficiente de actividad para una
deducción de esta expresión.
es adimensional
ya que la actividad no tiene dimensiones . Las actividades de los productos de
disociación están colocados en el numerador, las actividades de los reactivos
están colocadas en el denominador. Ver coeficiente de actividad para una
deducción de esta expresión.
Puesto
que la actividad es el producto de la concentración
y el coeficiente de actividad (γ) la definición
podría también escribirse como:
donde
[HA] representa la concentración de HA y Γ es un cociente de coeficientes de
actividad.

Variación del pKa
del ácido acético con la fuerza iónica.
Para
evitar las complicaciones que implica el uso de actividades, las constantes de
disociación se determinan,
cuando es posible, en un medio de alta fuerza iónica,
es decir, bajo condiciones en las que Γ se puede suponer que es siempre
constante. por ejemplo, el medio puede ser una solución de 0.1 M de nitrato de
sodio o 3 M de perclorato de potasio (1 M = 1 mol·dm−3,
una unidad de concentración molar). Además, en todos, salvo
en las soluciones más concentradas se puede suponer que la concentración de
agua, [H2O], es constante, aproximadamente 55 mol·dm−3.
Dividiendo K por los términos constantes y
escribiendo [H+] para la concentración de hidronio, se obtiene la
expresión:

Esta
es la definición de uso común. pKa se define como -log10Ka.
Nótese, sin embargo, que todos los valores de las constantes de disociación
publicados se refieren al medio iónico específico utilizado en su determinación
y que se obtienen diferentes valores con distintas condiciones, como se muestra
para ácido acético en la ilustración anterior.
Cuando las constantes publicadas se refieren a una fuerza iónica que no sea la
requerida para una aplicación particular, puede ajustarse mediante la teoría de
iones específicos (SIT) y otras teorías.10
Aunque
Ka parece tener la dimensión de concentración, de hecho, debe
ser a dimensional o no sería posible obtener su logaritmo.
La ilusión es el resultado de omitir el término constante [H_2O] de la
definición de la expresión. Sin embargo, no es inusual, sobre todo en textos
relativos a equilibrios bioquímicos, ver un valor citado con una dimensión
como, por ejemplo, "Ka = 300 M".
Ácidos
monopróticos
Variación del porcentaje (%)
de formación de un ácido monoprótico, AH, y su base conjugada, A-,
con la diferencia entre el pH y el pKa del ácido.
Después
de modificar la expresión que defineKa tomando logaritmos y
poniendo pH =-log 10 [H+] se obtiene:

Esta
es una forma de la ecuación de Henderson-Hasselbalch,
de la que pueden obtenerse las siguientes conclusiones:
- A
mitad de la neutralización [AH]/[A-] = 1; dado que log (1) = 0,
el pH a mitad de la neutralización es numéricamente igual al pKa.
Inversamente, cuando el pH = pKa, la concentración de AH
es igual a la concentración de A-.
- La
región tamponada se extiende sobre el
intervalo aproximado pKa ± 2, aunque el tamponamiento es
débil fuera del rango de pKa ± 1. Cuando pKa
± 1, entonces [AH]/[A->/sup>] = 10 o 1/10.
- Si
se conoce el pH, se puede calcular la relación [AH]/[A-]. Esta relación es
independiente de la concentración analítica del ácido.
En
el agua, los valores medibles de pKa varían en un rango que
va desde alrededor de -2 para un ácido fuerte a cerca de 12 para un ácido muy
débil (o base fuerte). Todos los ácidos con un valor de pKa
menores que -2 están disociados más del 99% a un pH de 0 (1 M de ácido). Esto
se conoce como efecto nivelador del disolvente ya que todos los ácidos parecen
estar al mismo nivel de ácidos fuertes, independientemente de sus
valores de pKa. Asimismo, todas las bases con un valor de pKa
mayor que el límite superior están desprotonadas en más del 99% para todos los
valores posibles de pH y se clasifican como bases fuertes.3
Un
ejemplo de ácido fuerte es el ácido clorhídrico, HCl, que tiene un valor de pKa,
estimado a partir de medidas termodinámicas de -9,3 en agua.11
La concentración de ácido no disociado en una solución 1 mol·dm−3
sería menor del 0.01% de las concentraciones de los productos de disociación.
Se dice que el ácidoclorhídrico está completamente disociado en solución
acuosa porque la cantidad de ácido no disociado es imperceptible. Cuando se
conocen el pKa y la concentración analítica del ácido, se
puede calcular fácilmente la extensión de la disociación y el pH de una
solución de un ácido monoprótico utilizando una tabla ICE (concentraciones iniciales,
cambio y concentraciones en el equilibrio).
Un
tampón de un pH deseado puede prepararse como
una mezcla de un ácido débil y su base conjugada. En la práctica, la mezcla
puede crearse disolviendo el ácido en agua, y agregando la cantidad necesaria
de ácido o base fuerte. El pKa del ácido debe ser inferior a
dos unidades de diferencia del pH de destino.
Ácidos
polipróticos
Porcentaje de formación de
especies en función del pH.
Porcentaje de formación de
especies calculada con el programa HySS para una solución 10 milimolar de ácido
cítrico. pKa1=3.13, pKa2 = 4.76, pKa3=6.40.
Los
ácidos polipróticos son ácidos que pueden perder más de un protón. La constante
de disociación para el primer protón puede indicarse como Ka1
y las constantes de disociación de los sucesivos protones como Ka2,
etc. El ácido fosfórico, H3PO4,
es un ejemplo de ácido poliprótido que puede perder tres protones.
equilibrio
|
Valor de pKa
|
H3PO4
|
pKa1 =
2.15
|
H2PO4−
|
pKa2 =
7.20
|
HPO42−
|
pKa3 =
12.37
|
Cuando
la diferencia entre valores sucesivos de pK es de alrededor de cuatro o
más unidades, como en este ejemplo, cada especie puede considerarse como un
ácido por derecho propio. De hecho, las sales de H2PO4− pueden cristalizarse
de una solución ajustando el pH alrededor de 5.5 y las sales de HPO42−
pueden cristalizarse de la solución ajustando el pH alrededor de 10. El
diagrama de distribución de especies muestra que las concentraciones de los dos
iones son máximas a pH 5.5 y 10.
Cuando
la diferencia entre valores sucesivos de pK es menor que aproximadamente
cuatro hay superposición entre el rango de pH de existencia de las especies en
equilibrio. A menor diferencia, mayor solapamiento. El caso del ácido cítrico
se muestra a la derecha; las soluciones de ácido cítrico están tamponadas en el
rango completo de pH de 2,5 a 7,5.
En
general, es cierto que los sucesivos valores de pK aumentan (Primera
regla de Pauling). por ejemplo, para un ácido diprótico, H2A, los dos equilibrios son:
H2A
HA−
+ H+
HA−
A2−
+ H+
se
puede observar que el segundo protón se retira de una especie cargada
negativamente. Dado que el protón tiene una carga positiva se necesita un
trabajo extra para arrancarlo; esta es la causa de la tendencia señalada
anteriormente. Los (anteriores) valores para el ácido fosfórico ilustran esta
regla, al igual que los valores del ácido vanádico, H3VO4.
Cuando se encuentra una excepción a la regla indica que está ocurriendo un
cambio importante en la estructura. En el caso de VO2+
(aq), el vanadio tiene geometría molecular octaédrica,
(número de coordinación 6), mientras que el ácido vanádico tiene geometría molecular tetraédrica,
(número de coordinación 4). Esta es la explicación de porqué pKa1
> pKa2 para los oxoácidos de vanadio (V).
equilibrio
|
Valor de pKa
|
[VO2(H2O)4]+
|
pKa1 =
4.2
|
H3VO4
|
pKa2 =
2.60
|
H2VO4−
|
pKa3 =
7.92
|
HVO42−
|
pKa4 =
13.27
|
Autoionización
del agua .
El
agua tiene propiedades tanto ácidas como básicas. La constante de equilibrio
para el equilibrio:
2 H2O
OH−
+ H3O+
viene
dada por:

Cuando,
como suele ser usual, la concentración de agua se puede considerar constante,
esta expresión puede reemplazarse por:

El
valor de Kw en condiciones estándar es
1.0×10−14. La constante de auto ionización del agua, Kw,
es por tanto, un caso especial de constante de disociación ácida.
Bases
Históricamente,
la constante de equilibrio Kb para una base se ha definido
como la constante de asociación para la protonación de la base, B, a la
forma ácida conjugada, HB+. Normalmente se obtiene a partir del
valor de Ka.
B +
H2O HB+
+ OH−
Utilizando
un razonamiento similar al usado anteriormente:
En
agua, la concentración del ion hidróxido,
[OH−], se relaciona con la concentración del ion hidrógeno mediante Kw
= [H+] [OH−], por tanto:

La
sustitución de la expresión de [OH−] en la expresión de Kb
da:

Cuando
Ka, Kb y Kw se determinan
en las mismas condiciones de temperatura y fuerza iónica, se sigue, tomando logaritmos
que pKb = pKw - pKa. En
solución acuosa a 25 °C, pKw es 13.9965,14
así pKb ~ 14 - pKa. En efecto, no hay
necesidad de definir pKb de forma separada de pKa,
pero se hace aquí porque los valores de pKb pueden
encontrarse en la literatura antigua.
Dependencia
de la temperatura
Todas
las constantes de equilibrio varían con la temperatura
de acuerdo con la ecuación de van't Hoff15

Acidez en soluciones no acuosas
Un
disolvente es más probable que promueva la ionización de una molécula ácida
disuelta en las siguientes circunstancias.16- Si
es un disolvente prótico,
capaz de formar enlaces de hidrógeno.
- Si
tiene un alto número donador, por
lo que es una base de Lewis fuerte.
- Si
tiene una alta constante dieléctrica (permitividad
relativa), por lo que es un buen disolvente de especies iónicas.
|
Propiedades de los
disolventes a 25oC
|
||
|
Disolvente
|
Número donador16
|
Constante dieléctrica16
|
|
Acetonitrilo
|
14
|
37
|
|
Dimetilsulfóxido
|
30
|
47
|
|
Agua
|
18
|
78
|
|
Valores de pKa
de ácidos
|
|||
|
HA
|
ACN
|
DMSO
|
agua
|
|
ácido p-Toluenosulfónico
|
8.5
|
0.9
|
fuerte
|
|
2,4-Dinitrofenol
|
16.66
|
5.1
|
3.9
|
|
Ácido Benzoico
|
21.51
|
11.1
|
4.2
|
|
Ácido Acético
|
23.51
|
12.6
|
4.756
|
|
29.14
|
18.0
|
9.99
|
|
|
BH+
|
|||
|
Pirrolidina
|
19.56
|
10.8
|
11.4
|
|
Trietilamina
|
18.82
|
9.0
|
10.72
|
|
1,8-Bis(dimetilamino)naftaleno{{
|11}}
|
18.62
|
7.5
|
12.1
|
|
Piridina
|
12.53
|
3.4
|
5.2
|
|
Anilina
|
10.62
|
3.6
|
9.4
|
HCl
+ CH3CO2H  Cl−
+ CH3C(OH)2+
Cl−
+ CH3C(OH)2+
ácido
+ base base
conjugada + ácido conjugado
Compare
esta reacción con lo que ocurre cuando el ácido acético se disuelve en el
disolvente más acídico ácido sulfúrico puro22
H2SO4
+ CH3CO2H HSO4−
+ CH3C(OH)2+
La
especie poco CH3C(OH)2+ es estable en estos
entornos. Para soluciones acuosas la función de acidez más
adecuada es la escala de pH]]. Se han propuesto otras funciones de acidez para medios no acuosos, la más
notable es la función de acidez de Hammett, H0,
para medios supe ácidos y su versión modificada H− para
medios superbásicos.
Dimerización de un ácido
carboxílico.
En
disolventes apróticos, se pueden formar oligómeros
por enlace de hidrógeno, como el conocido dímero del ácido acético. Un ácido también
puede formar enlaces de hidrógeno con su base conjugada. Este proceso, conocido
como homoconjugación, tiene el
efecto de aumentar la acidez de los ácidos, reduciendo sus valores efectivos de
pKa, mediante la estabilización de la base conjugada. La
homoconjugación aumenta en un factor de casi 800 el poder donador de protones
del ácido toluenosulfónico en una solución de acetonitrilo. En soluciones acuosas, la homoconjugation no se produce, porque el agua forma
enlaces de hidrógeno más fuertes a la base conjugada que al ácido.
Mezcla
de disolventes
Cuando
un compuesto tiene una solubilidad limitada en agua es una práctica corriente
(en la industria farmacéutica, por ejemplo) determinar los valores de pKa
en una mezcla de disolventes tal como agua/dioxano
o agua/metanol,
en las que el compuesto es más soluble.27
En el ejemplo que se muestra a la derecha, el valor de pKa
aumenta bruscamente con el porcentaje creciente de dioxano al disminuir la
constante dieléctrica de la mezcla.Un valor de pKa obtenido en una mezcla de disolventes no puede utilizarse directamente para soluciones acuosas. La razón de esto es que cuando el disolvente está en su estado estándar su actividad se “define” como la unidad. Por ejemplo, el estado estándar del agua: dioxano 9:1 es precisamente la mezcla disolvente, sin solutos añadidos. Para obtener el valor de pKa para utilizar en disoluciones acuosas tienen que ser extrapolados a una concentración de codisolvente de cero a partir de los valores obtenidos de varias mezclas de codisolvente.
Estos hechos quedan ocultados por la omisión del disolvente en la expresión que se utiliza normalmente para definir el pKa pero los valores de pKa obtenidos en una determinada mezcla de disolventes dada se puede comparar con cualquier otra, dando las fuerzas ácidas relativas. Lo mismo es cierto para los valores de pKa obtenidos en un disolvente no acuoso particular tal como el DMSO.
Hasta el momento actual, no se ha desarrollado una escala de constantes de disociación universal e independiente del disolvente, ya que no se conoce la forma de comparar los estados estándar de dos disolventes diferentes.